![[GIRI]](/global/girilogo.png)
Education
TE identification and mapping
Given the huge diversity of TEs in most eukaryotic genomes and the lack of sequence conservation across species, it is often a challenge to mine TE sequences from a genome (mapping or detection). To some extent, this task can be done with blasting against a reference TE library (Repbase database) by Censor or RepeatMasker. For new genome sequences, it is necessary to perform TE mining or discovery from a scratch. A plenty of computing tools have been developed for this goal. Based on the underlying approaches, these tools can be divided into three categories. The first group are based on the homology or repetitiveness of TEs, such as RepeatScout and Recon. The second group are based on the structure features of certain types of TEs. The third group are based on comparative genomic method. Different tools have their own limits in performance. Some efforts attempt to combine multiple approaches, such as REPET and RepeatModeler. Up to now, a common problem of these automate tools is that a significant number of the output sequences are flawed in sequence accuracy and annotation. Manual curation is often required to get the full-length, high-quality family consensus. The workflow adopted in Repbase is illustrated below.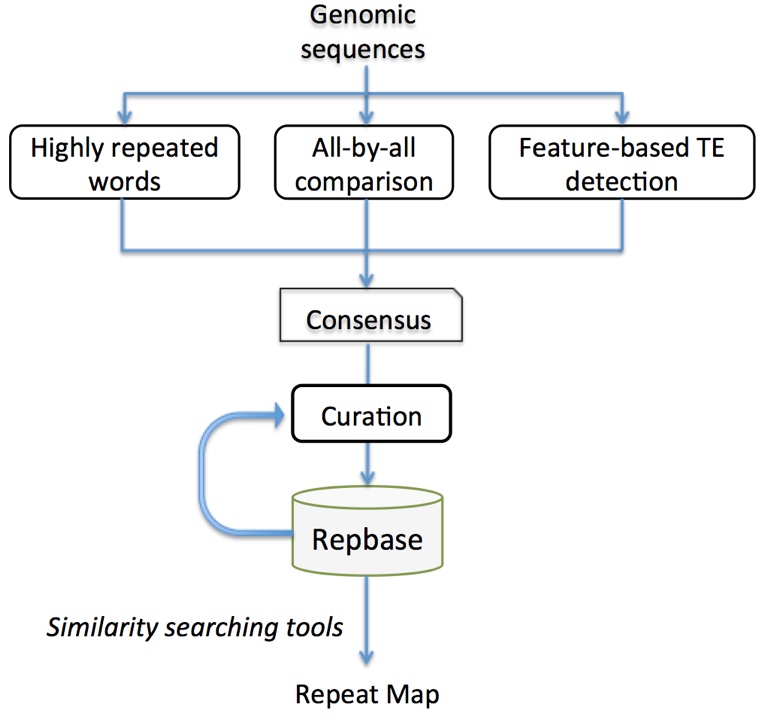
Further readings
- Price, AL, NC Jones, and PA Pevzner (2005), 'De novo identification of repeat families in large genomes.', Bioinformatics, 21 Suppl 1 i351–8.
- Bao, Z and SR Eddy (2002), 'Automated de novo identification of repeat sequence families in sequenced genomes.', Genome Res, 12 (8), 1269–76.
- Bergman, CM and H Quesneville (2007), 'Discovering and detecting transposable elements in genome sequences.', Brief Bioinform, 8 (6), 382–92.
- Flutre, T, et al. (2011), 'Considering transposable element diversification in de novo annotation approaches.', PLoS One, 6 (1), e16526.
Weidong Bao, Ph. D.